

La sindrome riproduttiva e respiratoria dei suini (PRRS) rimane di gran lunga la malattia con il maggiore impatto economico nel settore suinicolo ed il suo controllo è tutt'altro che soddisfacente. Un quadro migliore e più completo della variazione del virus della PRRS e del monitoraggio della circolazione di nuovi ceppi all'interno di una determinata area / paese / continente aiuterebbe sicuramente i veterinari ed i produttori ad implementare programmi di controllo ed eventualmente di eradicazione. Per questo motivo, dalla fine del 1990, il sequenziamento del virus della PRRS ha iniziato ad essere disponibile in tutto il mondo, principalmente in Nord America, Europa e Sud-Est asiatico.
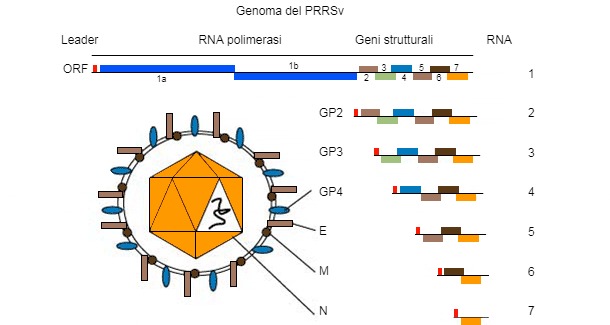
Il genoma del virus della PRRS (immagine 1) consiste in una molecola di RNA a filamento singolo che lo rende incline a "fare errori" (mutazioni genetiche) durante la sua replica nell'ospite. Questa "tendenza a sbagliare" comporta la presenza sul campo di diversi ceppi di virus della PRRS, tutti unici nella loro sequenza genetica. C'è ancora un dibattito tra veterinari di campo e ricercatori se queste differenze nelle sequenze contribuiscono a "comportamenti diversi" (siano essi clinico-patologici o immunologici).

Concetti di base del sequenziamento del virus della PRRS
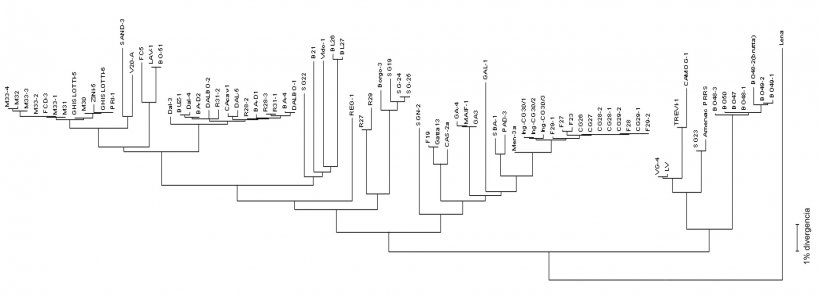
Il sequenziamento del virus viene eseguito da prodotti di PCR provenienti da campioni di campo (sieri, tessuti, fluidi orali) ottenendo la lettura di nucleotidi generalmente da alcuni frammenti del genoma dell'RNA virale (vedi figura 2) in alcune regioni target - ORF (Open Reading Frame) e quindi confrontare la percentuale di omologia attraverso l'analisi filogenetica eseguita utilizzando un software specifico. Il risultato di questo processo fornisce il grado di somiglianza (omologia) tra i diversi ceppi di virus PRRS. Utilizzando il software di visualizzazione grafica, è anche possibile ottenere un dendrogramma (o "albero filogenetico") che mostra il grado di relazione con la sequenza del virus di riferimento (vedere la figura 3).

Il genoma del PRRSV codifica almeno 10 ORF. I più comunemente usati per il sequenziamento - sebbene rappresentino rispettivamente solo il 4% e il 3% del genoma completo, sono ORF5 (che codifica per la proteina non glicosilata E) e ORF7 (che codifica per la proteina nucleocapsidica (N)). ORF5 rappresenta una regione più variabile, mentre ORF7 rappresenta una regione più conservata. A causa di ciò, lo stesso grado di variazione (ad esempio una variazione del 5%) trovato in ORF7 è più "drammatico" in termini di cambiamento genetico, rispetto alla stessa variazione in ORF5. L'interpretazione delle somiglianze (cioè se i virus sono o meno collegati) richiede molte più informazioni aggiuntive, poiché la velocità del cambiamento genetico può essere molto variabile.
È estremamente importante mantenere un file di registrazione di tutte le sequenze identificate in modo univoco, prestando attenzione alla data, al tipo di azienda (sito 1-2-3), al flusso dei suini, alla posizione (latitudine / longitudine GPS) e all'origine della sequenza (tipo di animale / tessuto / campione). Ad oggi, il nostro database di sequenze di virus PRRS copre oltre 1.300 sequenze di ORF7 dal 2002. Per interpretare e dare un senso alle differenze, è ancora più importante correlare le singole sequenze agli eventi clinici come il numero di scrofe che abortiscono ed alla mortalità pre-svezzamento nei Siti 1 o il tasso di mortalità nei siti 2 e 3.
Domande pratiche
Le domande frequenti degli allevatori e veterinari sono:
- Le differenze genetiche osservate tra le sequenze rappresentano la variazione normale di un singolo ceppo di PRRS in un'azienda / sistema, oppure rappresentano più ceppi diversi presenti nello stesso momento, o in uno spazio temporale breve, in un'azienda / sistema?
- Quello che sto avendo ora è un "nuovo focolaio" causato da un "nuovo ceppo" o è un ricircolo?
Per rispondere a queste domande dobbiamo concordare sul grado accettato di omologia di due ceppi virali raccolti entro un certo periodo di tempo (12-24 mesi?). In altre parole, il "punto di taglio" della somiglianza. Un'omologia del 97-98% nella sequenza (o una differenza del 2-3%) è un valore generalmente accettato. Secondo la mia esperienza, è abbastanza difficile vedere un cambiamento superiore al 2% in una "popolazione chiusa clinicamente stabile" (una popolazione convenzionale di scrofe o un flusso di suini) poiché quello che osserviamo è che otteniamo lo "stesso ceppo" per un periodo fino a 3 anni in una singola popolazione clinicamente stabile. Al polo opposto, ogni volta che notiamo un'attività consistente della PRRS, viene recuperato un ceppo "nuovo" e filogeneticamente diverso (omologia del 90% o inferiore). Sfortunatamente, non sappiamo con certezza se queste grandi differenze che a volte osserviamo siano il risultato di un improvviso cambiamento nel virus / mutazione (improbabile secondo la mia opinione personale) o dell'introduzione di un nuovo ceppo. Ciò che è chiaro e ben accettato è il fatto che la somiglianza / diversità genetica non è in alcun modo predittiva della somiglianza immunologica (cioè, indicativa di immunità crociata protettiva) e non è affatto predittiva della patogenicità intrinseca (non dice se un particolare ceppo è "buono" o "cattivo").
Le sequenze complete del genoma attualmente disponibili (purtroppo ancora di più per scopi di ricerca piuttosto che per uso diagnostico quotidiano) aiuteranno sicuramente a rispondere a questa domanda.
È molto importante analizzare le nuove sequenze di virus PRRS con un ampio set di riferimento che rappresenta l'allevamento, il sistema e la regione, nonché le sequenze di vaccini commerciali disponibili (ciò consentirà di distinguere tra ceppi di campo e di vaccino). In questo momento stiamo ancora utilizzando un software open source gestito dall'Università di Padova per costruire i nostri alberi filogenetici e tenerli organizzati dal flusso dei suini nel totale del nostro sistema di produzione; nel prossimo futuro potremmo unirci ad altri due programmi informatici "ad hoc" (Bioportal dell'Università di Davis-California e CLASSIFARM-PATH di IZSLER (Brescia, Italia) che avranno una serie molto più ampia di sequenze da confrontare e consentire una migliore comprensione del virus della PRRS circolante in Italia e possibilmente nell'UE.
Ringraziamenti: Grazie al Prof. Michele Drigo (UNI-PD) per l'interessante discussione e revisione di questo documento.




