Introduzione
Il genoma del virus della sindrome riproduttiva e respiratoria suina (PRRSV) è formato da una catena semplice di RNA, il che lo fa molto predisposto alle mutazioni genetiche. Questo fa sì che ogni ceppo di PRRSV sia unico, pertanto la tipizzazione genetica è un metodo utile per la diagnostica e per il controllo della malattia. La diagnostica attraverso la tipizzazione genetica si fa determinando l'ordine dei nucleotidi all'interno di una coppia di DNA da un frammento del genoma RNA – mediante la sequenziazione del DNA. Attualmente il frammento più utilizzato è l'ORF5, il gene che codifica per la glicoproteina più importante della copertura, principalmente perchè mostra una grande diversità genetica.

Diagnostica per la sequenziazione del DNA
La discriminante tra il PRRSV tipo 1 (europeo) e tipo 2 (americano) si fa facilmente con la maggior parte delle PCR diagnostiche, però la differenziazione tra i ceppi individuali all'interno di ognuno dei due genotipi, richiede la sequenziazione del DNA. Per questo, si usano i fluidi corporei o i tessuti, con una carica virale tra il moderato e importante del PRRSV, dai quali si isola l'RNA, che è accoppiato al DNA, mediante trascrizione inversa; pertanto si amplifica il gene ORF5 mediante la PCR e si invia ad un laboratorio di sequenziazione del DNA. Questo processo è molto automatizzato e richiede tra 1 e 3 giorni. I dati grezzi della sequenziazione si mandano al laboratorio diagnostico per le loro analisi. Il report normalmente include la sequenza dei nucleotidi del ceppo, la sua similitudine a ceppi vaccinali standard ed alcuni laboratori forniscono una comparazione con un pannello di riferimento standard dei ceppi isolati selvaggi di PRRSV in forma di dendrogramma o la comparazione col data base dei PRRSV di un sistema di produzione.
Analisi delle sequenziazioni del PRRSV
La similitudine o l'identicità, tra sequenziazioni si determina allineando 2 o più sequenze utilizzando un programma informatico. La figura 1 rappresenta un esempio di allineamento di sequenziazioni di ORF5 di 5 allevamento differenti. Le comparazioni per coppie delle percentuali di identità vengono mostrate nella tabella. Le identità oscillano tra 81,2 e 99,8 %. Il dendrogramma che ne deriva dall'analisi filogenetica mostra un raggruppamento delle sequenziazioni similari (figura 2). Una questione chiave per i produttori ed i veterinari è se le differenze genetiche osservate tra sequenze rappresentano una variazione normale di uno stesso ceppo di PRRSV dell'allevamento o rappresentano vari ceppi distinti nello stesso allevamento.

Figura 1: Frammento dell'allineamento delle sequenze ORF5 di ceppi di PRRSV di 5 allevamenti distinti. Degli allevamenti 1, 4 e 5, si ottennero molteplici sequenze. I punti rappresentano posizioni identiche al ceppo di referenza: un PRRSV tipo 1, Lelystad.
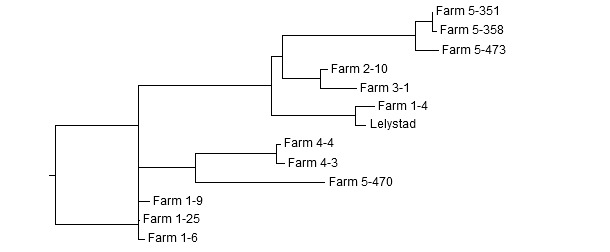
Figura 2: Dendrogramma di sequenze ORF5 ottenute da 5 allevamenti distinti. Esempio di interpretazione: nell'allevamento 1 ci sono 2 ceppi non correlati. Tre sequenze sono > 99 % identiche tra loro, mentre la quarta è solo correlata un ~ 83 %. In cambio, sta correlata con il virus Lelystad. I ceppi dell'allevamento 2 e 3 sono molto correlati (98,2 % identici). Due ceppi dell'allevamento 4 sono molto correlati (99,5 % identici).Nell'allevamento 5 ci sono 2 ceppi distinti: 3 sequenze sono > 98 % identiche tra loro e 81 % con la quarta.
Tab.: Percentuale di identità per coppie tra tutte le sequenze allineate di ORF5 di una serie di campioni di PRRSV tipo 1.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83,5 | 83,3 | 83,7 | 93,2 | 92,6 | 86,1 | 86,0 | 88,1 | 88,0 | 83,2 | 88,3 | 98,7 | 1-4 |
| *** | 99,2 | 99,7 | 84,2 | 83,0 | 86,0 | 86,0 | 81,4 | 81,2 | 86,3 | 81,4 | 82,1 | 1-6 | |
| *** | 99,5 | 84,5 | 83,3 | 85,6 | 85,6 | 81,4 | 81,2 | 86,5 | 81,4 | 81,9 | 1-9 | ||
| *** | 84,5 | 83,3 | 86,0 | 86,0 | 81,7 | 81,5 | 86,8 | 81,7 | 82,2 | 1-25 | |||
| *** | 98,2 | 86,6 | 86,5 | 91,1 | 90,9 | 84,2 | 90,4 | 93,3 | 2-10 | ||||
| *** | 84,4 | 84,2 | 90,2 | 90,0 | 83,3 | 89,7 | 92,9 | 3-1 | |||||
| *** | 99,5 | 82,5 | 82,7 | 90,6 | 82,5 | 84,8 | 4-4 | ||||||
| *** | 82,3 | 82,5 | 90,9 | 82,2 | 84,6 | 4-3 | |||||||
| *** | 99,8 | 81,0 | 98,3 | 88,4 | 5-351 | ||||||||
| *** | 81,2 | 98,2 | 88,2 | 5-358 | |||||||||
| *** | 80,9 | 83,2 | 5-470 | ||||||||||
| *** | 88,8 | 5-473 | |||||||||||
| *** | Lelystad |
Interpretazione delle sequenze del PRRSV
La sequenza dell'ORF5 ha circa 600 nucleotidi. Diverse stime indicano che la percentuale globale di mutazione di questo gene è circa dello 0,5 - 1 % all'anno. Le variazioni nella percentuale di cambio genetico sono determinate da diversi fattori non virali. Il livello di immunità specifica nei confronti del PRRSV dei suini ha un grande impatto sulla replicazione e trasmissione virale, esercitando una forte pressione inibitrice che diminuisce il numero di coppie. Una carica virale ridotta comporta una minore percentuale di trasmissione, limitando tuttavia maggiormente la replicazione virale e diminuendo la percentuale di variazione. Lo stesso virus può mostrare diversi tassi di variazione genetica in condizioni particolari e pertanto, in alcuni casi, si possono osservare tassi di variazione maggiori o minori del range suggerito dallo 0,5 - 1% all'anno.
La questione principale nell'analisi genetica è sapere se 2 sequenze sono molto correlate (appartengono a 2 ceppi correlati dello stesso ceppo) o sono indipendenti (appartengono a 2 ceppi non correlati). Comunemente si accetta che 2 ceppi isolati di PRRSV sono correlati o no se la loro similitudine è al di sopra o al di sotto del 97 - 98 %. E' evidente, che basarsi unicamente su una differenza genetica di un 2 o 3 % tra 2 ceppi isolati, senza alcuna informazione aggiuntiva, può portare a conclusioni errate. Le differenze tra varianti di uno stesso ceppo circolante in una popolazione durante molti anni, possono eccedere questa cifra. L'interpretazione della relazione sarà maggiore se si ha accesso ad informazioni aggiuntive, includendo le date ed i luoghi degli isolamenti. E' molto importante comparare le nuove sequenze di PRRSV con un ampio gruppo di referenza che sia rappresentativo dell'allevamento, del sistema e della regione e che rappresenti anche la diversità genetica globale.
La sequenziazione del DNA delle catene del PRRSV, può indicare una correlazione di vicinanza, o di indipendenza, delle catene(figura 2), però non può predire o spiegare la protezione immunologica o gli episodi di PRRS in allevamenti immuni. Nè permette la predizione dell'evoluzione clinica dell'infezione di un determinato ceppo, dato che i marcatori genetici di virulenza, purtroppo, non sono ancora stati identificati.
Attualmente, ci sono molti progetti regionali in corso per il controllo o eliminazione del PRRS, specialmente negli USA, però anche in Europa. Tenere una visione completa della variabilità virale all'inizio del progetto per il controllo e l'eliminazione della PRRS, è essenziale per un efficace controllo dei progressi, dell'incisività dei processi implementati e per identificare i nuovi virus introdotti nell'allevamento dalle zone confinanti.